Advanced usage
This tutorial demonstrates a workflow of state-of-the-art analyses using single-molecule fluorescence movies in living cells.
To demonstrate single-molecule analysis using Slitflow, we used live-cell single-molecule movies of RNA polymerase II—that is, Pol II, a transcriptional protein complex—from cultured cells. Human U2OS cells expressing Halo-RPB1—the largest subunit of Pol II labeled with a self-labeling HaloTag—were stained with 3 nM Janelia Fluor fluorescence substrate, and excited using HILO illumination. 300 frames were captured at 33.33 ms per frame using an EMCCD camera. Single-molecule movies were used to extract and track bright spots and to visualize and analyze the trajectory on a single pipeline script.
Additional installation
This tutorial requires additional python packages. Please install through the pip command as follows. fastspt should be installed from the forked repository.
pip install trackpy tramway git+https://gitlab.com/yumaitou/Spot-On-cli.git@py310
The current version of original fastspt installed from PyPI has an error regarding the version file while importing a package in Python 3.10 environment. The modified version of fastspt is temporally available from the forked repository.
Caution
Trackpy package requires installation of Visual Studio 2008 c++ runtime. Please install appropriate version for your PC from microsoft website.
Download movies
You can download image data of single-molecule movies from zenodo
as a zip file (142 MB). Please unzip the file outside of the project
directory. In this tutorial, we will assume that the data are unzipped in
the slitflow/data directory in the user home directory.
Import movies
The image data are assumed to be stored in the slitflow/data directory in your
home directory. The script below loads single-molecule movies, mask images
of cell nuclei, and the parameter CSV file.
# make a project directory (in the user directory)
prj_dir = os.path.join(
os.path.expanduser("~"), "slitflow", "getting_started_advanced")
if not os.path.isdir(prj_dir):
os.makedirs(prj_dir)
print(prj_dir)
PL = sf.manager.Pipeline(prj_dir)
# path to downloaded image data directory
raw_dir = os.path.join(os.path.expanduser("~"), "slitflow",
"data", "getting_started_advanced")
pitch = 0.0710837445886793 # [um/pix]
interval = 0.03333 # [s]
for i in [1, 2, 3]:
path = os.path.join(raw_dir, "rpb1", "rpb1-" + str(i) + ".tif")
PL.add(sf.load.tif.SplitFile(), 0, (1, 1), "rpb1", "raw",
["RPB1"], [], [],
{"path": path, "length_unit": "um", "pitch": pitch,
"interval": interval, "value_type": "uint8", "indexes": [i],
"split_depth": 1})
path = os.path.join(raw_dir, "mask", "mask.tif")
PL.add(sf.load.tif.SingleFile(), 0, (2, 1), "mask", "raw",
["RPB1"], [], [],
{"path": path, "length_unit": "um", "pitch": pitch,
"value_type": "uint8", "split_depth": 0})
PL.save("pipeline_1_load")
PL.run()
Tracking
Single-molecule tracking requires pre-processing and tracking algorithms that are appropriate for the characteristics of the acquired images. Here, we implemented a multistep customized process that focused on improving the location accuracy and processing time.
First, fluorescent spots were detected using a Difference of Gaussian filter and the local maximum—as used in u-track and TrackMate —and then selected using a cell nucleus region mask and an intensity threshold. The positions were further refined by 2D Gaussian fitting using a scipy.optimize.curve fit, the trajectories being extracted using the link function of Trackpy. To exclude noise trajectories, those with at least nine steps were selected.
These processes can be executed using the following pipeline script.
prj_dir = os.path.join(
os.path.expanduser("~"), "slitflow", "getting_started_advanced")
PL = sf.manager.Pipeline(prj_dir)
PL.add(sf.img.filter.DifferenceOfGaussian(), 3, (1, 2), None, "dog",
["RPB1"], [(1, 1)], [2], {"wavelength": 0.6, "NA": 1.4, "split_depth": 1})
PL.add(sf.img.filter.LocalMax(), 3, (1, 3), None, "localmax",
["RPB1"], [(1, 2)], [2], {"split_depth": 1})
PL.add(sf.loc.convert.LocalMax2Xy(), 3, (1, 4), None, "xy",
["RPB1"], [(1, 3)], [2], {"split_depth": 1})
PL.add(sf.loc.mask.BinaryImage(), 2, (1, 5), None, "mask",
["RPB1"], [(1, 4), (2, 1)], [1, 1], {"split_depth": 1})
PL.add(sf.tbl.filter.CutOffPixelQuantile(), 2, (1, 6), None, 'cutoff',
["RPB1"], [(1, 5)], [2], {"calc_col": "intensity", "cut_factor": 4, "split_depth": 1})
PL.add(sf.loc.fit.Gauss2D(), 3, (1, 7), None, 'refine',
["RPB1"], [(1, 1), (1, 6)], [2, 2], {"half_width": 4, "split_depth": 1})
PL.add(sf.trj.wtrackpy.Link(), 3, (1, 8), None, 'trj',
["RPB1"], [(1, 7)], [1], {"search_range": 0.8, "split_depth": 1})
PL.add(sf.trj.filter.StepAtLeast(), 2, (1, 9), None, 'long',
["RPB1"], [(1, 8)], [1], {"step": 9, "group_depth": 2, "split_depth": 1})
PL.add(sf.tbl.math.Centering(), 1, (1, 10), None, "center",
["RPB1"], [(1, 9)], [1],
{"calc_cols": ["x_um", "y_um"], "group_depth": 1, "split_depth": 1})
PL.save("pipeline_2_tracking")
PL.run()
The first three processes can be replaced with
slitflow.loc.convert.LocalMax2XyWithDoG to reduce calculation time and
file size.
Since this strategy is just one example, you can customize the pipeline to suit the feature of images and the behavior of target molecules.
Show trajectory
The following pipeline creates the trajectory image for each cell nucleus.
prj_dir = os.path.join(
os.path.expanduser("~"), "slitflow", "getting_started_advanced")
PL = sf.manager.Pipeline(prj_dir)
PL.add(sf.fig.line.Simple(), 2, (3, 1), "show", "fig",
["RPB1"], [(1, 10)], [1],
{"calc_cols": ["x_um", "y_um"], "group_depth": 2, "split_depth": 1})
PL.add(sf.fig.style.Basic(), 2, (3, 2), None, "style",
["RPB1"], [(3, 1)], [1],
{"size": [4, 4], "margin": [0, 0, 0, 0],
"limit": [-14, 14, -14, 14], "tick": [[-15, 15], [-15, 15]],
"is_box": True, "line_widths": 0.7,
"split_depth": 1})
PL.add(sf.fig.figure.ToTiff(), 2, (3, 3), None, "tif",
["RPB1"], [(3, 2)], [1],
{"scalebar": [5, 0.05, 0.05, 2, [0, 0, 0]],
"dpi": 300, "split_depth": 0})
PL.add(sf.img.montage.RGB(), 0, (3, 4), None, "mtg",
["RPB1"], [(3, 3)], [0],
{"grid_shape": [1, 3], "padding_width": 0, "split_depth": 0})
PL.save("pipeline_3_show_trajectory")
PL.run()

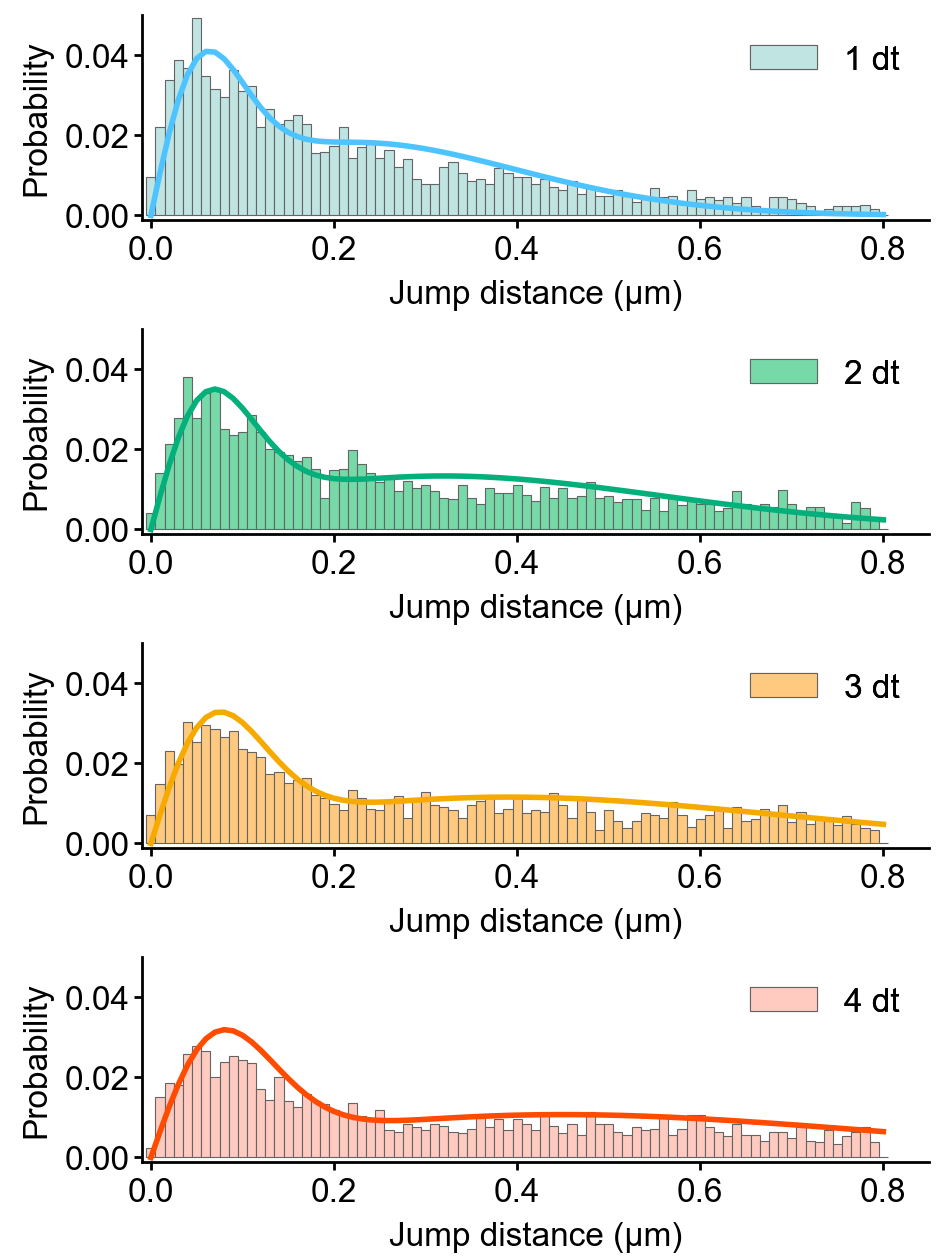
Spot-On analysis
Spot-On is state-of-the-art kinetic modeling of single particle trajectories (Hansen et al., 2017). Spot-On is provided as web-interface, python package, and MatLab backend.
Slitflow provides wrapping classes of the fastspt python package, including calculating jump length distribution, fitting the distribution with a model, and creating model curves.
The following example fits the jump length distribution of extracted trajectories with the two-component model with explicit localization error and without Z correction.
prj_dir = os.path.join(
os.path.expanduser("~"), "slitflow", "getting_started_advanced")
PL = sf.manager.Pipeline(prj_dir)
PL.add(sf.trj.wfastspt.JumpLenDist(), 0, (4, 1), "spoton", "hist",
["RPB1"], [(1, 9)], [0],
{"trj_depth": 2, "MaxJump": 0.8, "BinWidth": 0.01, "CDF": False,
"TimePoints": 5, "split_depth": 2})
PL.add(sf.trj.wfastspt.FitJumpLenDist2comp(), 0, (4, 2), None, "fit2",
["RPB1"], [(4, 1)], [0],
{"lower_bound": [0.05, 0.0001, 0], "upper_bound": [25, 0.08, 1],
"LocError": 0.035, "iterations": 3, "dZ": 0.700, "useZcorr": False,
"init": [0.5, 0.003, 0.3], "split_depth": 0})
PL.add(sf.trj.wfastspt.ModelJumpLenDist(), 0, (4, 3), None, "model",
["RPB1"], [(4, 1), (4, 2)], [0, 0],
{"show_pdf": True, "split_depth": 2})
PL.save("pipeline_4_spot_on")
PL.run()
This pipeline exports the resulting CSV files of each task, including jump length distributions, fitted parameters, and model curves.
Using the following pipeline, we can create the histogram images of the jump length distribution overlayed with the model curve.
prj_dir = os.path.join(os.path.expanduser(
"~"), "slitflow", "getting_started_advanced")
PL = sf.manager.Pipeline(prj_dir)
# path to figure style table
raw_dir = os.path.join(os.path.expanduser("~"), "slitflow", "data",
"getting_started_advanced")
path = os.path.join(raw_dir, "param", "spoton_fig.csv")
# all required Data should be split into fig unit
PL.add(sf.fig.bar.WithModel(), 2, (4, 4), None, "fig",
["RPB1"], [(4, 1), (4, 3)], [2, 2],
{"calc_cols": ["jump_dist", "prob"],
"model_cols": ["jump_dist", "prob"],
"group_depth": 2, "group_depth_model": 2, "split_depth": 2})
PL.add(sf.load.table.SingleCsv(), 0, (4, 5), None, "fig_param",
["RPB1"], [], [],
{"path": path, "col_info": [
[1, "is_cdf", "int32", "num", "Whether histogram is CD"],
[2, "dt", "int32", "num", "Time difference of jump step"],
[0, "legend", "str", "none", "Legend string"],
[0, "marker_colors", "str", "none", "Edge and face colors"],
[0, "line_colors", "str", "none", "Line colors"]],
"split_depth": 2})
PL.add(sf.fig.style.ParamTable(), 0, (4, 6), None, "fig_style",
["RPB1"], [(4, 4), (4, 5)], [2, 2],
{"size": [6, 2], "margin": [0.9, 0.6, 0.1, 0.1],
"label": ["Jump distance (\u03bcm)", "Probability"],
"is_box": True, "format": ["%.1f", "%.2f"],
"limit": [-0.01, 0.85, -0.001, 0.05],
"tick": [[0, 0.2, 0.4, 0.6, 0.8], [0, 0.02, 0.04]],
"marker_widths": 0.2})
PL.add(sf.fig.figure.ToTiff(), 0, (4, 7), None, "fig_tif",
["RPB1"], [(4, 6)], [1], {"split_depth": 0})
PL.add(sf.img.montage.RGB(), 0, (4, 8), None, 'fig_mtg',
["RPB1"], [(4, 7)], [0],
{"grid_shape": [4, 1], "padding_width": 0, "split_depth": 0})
PL.save("pipeline_5_spot_on_figure")
PL.run()
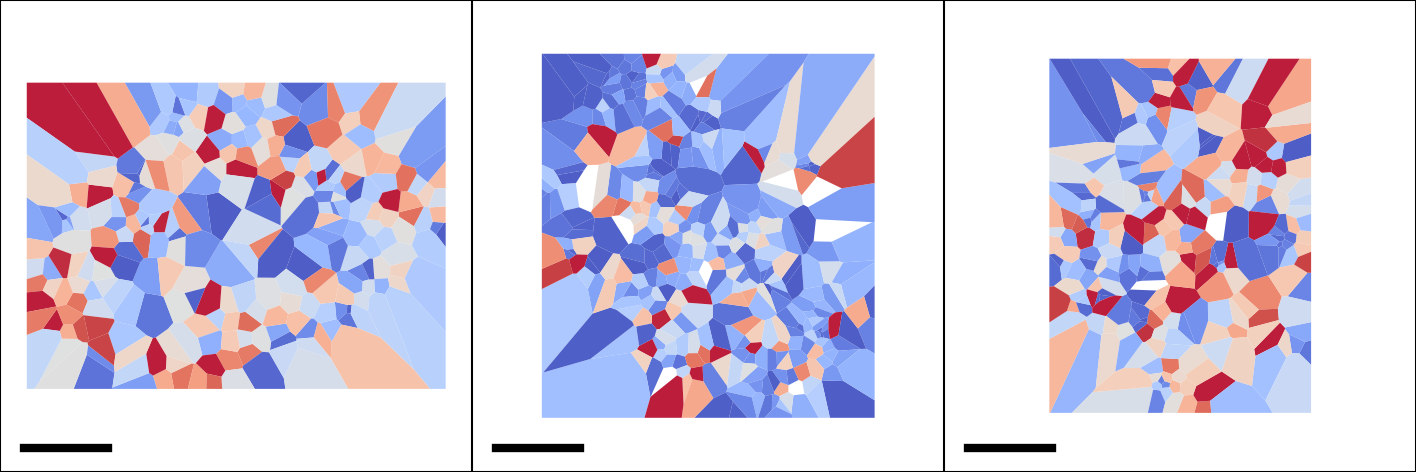
TRamWAy analysis
TRamWAy is a cutting-edge analysis tool for single molecule dynamics such as diffusivity and potential energy spatiotemporally. (Laurent et al., 2022). TRamWAy is provided as a python package tramway.
Slitflow provides wrapping classes of some of the helper functions in the tramway package, including tessellation, inference, and map_plot.
The following example calculates and visualizes the spatial map of molecular diffusivity for each cell nucleus.
prj_dir = os.path.join(
os.path.expanduser("~"), "slitflow", "getting_started_advanced")
PL = sf.manager.Pipeline(prj_dir)
PL.add(sf.trj.wtramway.Tessellation(), 1, (5, 1), "tram", "tess",
["RPB1"], [(1, 10)], [1], {"method": "gwr", "split_depth": 1})
PL.add(sf.trj.wtramway.Inference(), 0, (5, 2), None, "infer",
["RPB1"], [(5, 1)], [1], {"mode": "d"})
PL.add(sf.trj.wtramway.MapPlot(), 2, (5, 3), None, "map",
["RPB1"], [(5, 1), (5, 2)], [1, 1],
{"feature": "diffusivity", "param": {"unit": "std"}})
PL.add(sf.fig.style.Basic(), 0, (5, 4), None, "fig_style",
["RPB1"], [(5, 3)], [1],
{"size": [4, 4], "margin": [0, 0, 0, 0],
"limit": [-14, 14, -14, 14], "tick": [[-15, 15], [-15, 15]],
"clim": [0, 0.06], "cmap": "coolwarm"})
PL.add(sf.fig.figure.ToTiff(), 0, (5, 5), None, "fig_tif",
["RPB1"], [(5, 4)], [1],
{"scalebar": [5, 0.05, 0.05, 2, [0, 0, 0]],
"dpi": 300, "split_depth": 0})
PL.add(sf.img.montage.RGB(), 0, (5, 6), None, 'fig_mtg',
["RPB1"], [(5, 5)], [0],
{"grid_shape": [1, 3], "padding_width": 0, "split_depth": 0})
PL.add(sf.fig.style.ColorBar(), 0, (5, 7), None, "colorbar",
["RPB1"], [(5, 4)], [1],
{"tick": [0, 0.02, 0.04, 0.06], "format": "%0.2f"})
PL.add(sf.fig.figure.ToTiff(), 0, (5, 8), None, "cb_tif",
["RPB1"], [(5, 7)], [1], {"split_depth": 1})
PL.save("pipeline_6_tramway")
PL.run()
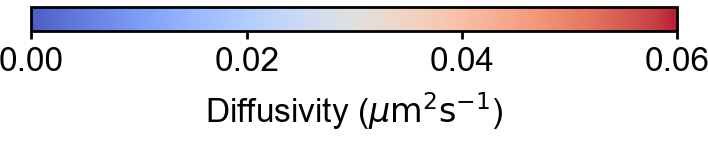
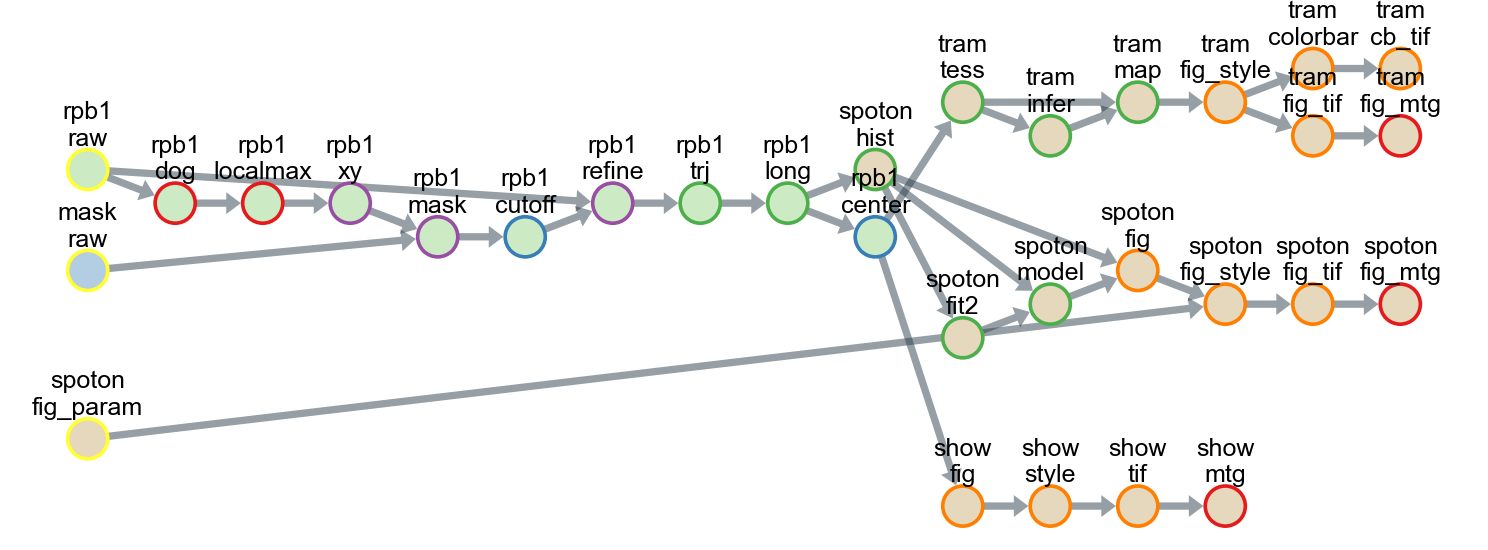
Make pipeline flowchart
All tasks, including tracking, analysis, and drawing, can be saved as a single pipeline script text file in the CSV format for reuse and distribution. Using the pipeline script, a series of data-processing steps from the raw data to the final image could be exported as a flowchart.
Each circle in the flowchart represents an individual task corresponding to an analysis subfolder in the project directory. The arrows between circles represent data dependencies. In this example, 26 different classes were used, and all the data were stored in 31 subfolders in five groups.
The flowchart can be created with the following script:
prj_dir = os.path.join(os.path.expanduser(
"~"), "slitflow", "getting_started_advanced")
PL = sf.manager.Pipeline(prj_dir)
PL.load(["pipeline_1_load", "pipeline_2_tracking", "pipeline_3_show_trajectory",
"pipeline_4_spot_on", "pipeline_5_spot_on_figure", "pipeline_6_tramway"])
PL.make_flowchart("pipeline", "grp_ana", scale=(0.6, 1.8))